ID:APOA1_HUMAN DESCRIPTION: RecName: Full=Apolipoprotein A-I; Short=Apo-AI; Short=ApoA-I; AltName: Full=Apolipoprotein A1; Contains: RecName: Full=Truncated apolipoprotein A-I; AltName: Full=Apolipoprotein A-I(1-242); Flags: Precursor; FUNCTION: Participates in the reverse transport of cholesterol from tissues to the liver for excretion by promoting cholesterol efflux from tissues and by acting as a cofactor for the lecithin cholesterol acyltransferase (LCAT). As part of the SPAP complex, activates spermatozoa motility. SUBUNIT: Interacts with APOA1BP and CLU. Component of a sperm activating protein complex (SPAP), consisting of APOA1, an immunoglobulin heavy chain, an immunoglobulin light chain and albumin. Interacts with NDRG1. INTERACTION: P05067:APP; NbExp=5; IntAct=EBI-701692, EBI-77613; SUBCELLULAR LOCATION: Secreted. TISSUE SPECIFICITY: Major protein of plasma HDL, also found in chylomicrons. Synthesized in the liver and small intestine. The oxidized form at Met-110 and Met-136 is increased in individuals with increased risk for coronary artery disease, such as in carrier of the eNOSa/b genotype and exposure to cigarette smoking. It is also present in increased levels in aortic lesions relative to native ApoA-I and increased levels are seen with increasing severity of disease. PTM: Palmitoylated. PTM: Met-110 and Met-136 are oxidized to methionine sulfoxides. PTM: Phosphorylation sites are present in the extracellular medium. MASS SPECTROMETRY: Mass=28081; Method=Electrospray; Range=25-267; Note=Without methionine sulfoxide; Source=PubMed:12576517; MASS SPECTROMETRY: Mass=28098; Method=Electrospray; Range=25-267; Note=With 1 methionine sulfoxide, oxidation at Met-110; Source=PubMed:12576517; MASS SPECTROMETRY: Mass=28095; Method=Electrospray; Range=25-267; Note=With 1 methionine sulfoxide, oxidation at Met-136; Source=PubMed:12576517; MASS SPECTROMETRY: Mass=28114; Method=Electrospray; Range=25-267; Note=With 2 methionine sulfoxides, oxidation at Met-110 and Met- 136; Source=PubMed:12576517; DISEASE: Defects in APOA1 are a cause of high density lipoprotein deficiency type 2 (HDLD2) [MIM:604091]; also known as familial hypoalphalipoproteinemia (FHA). Inheritance is autosomal dominant. DISEASE: Defects in APOA1 are a cause of the low HDL levels observed in high density lipoprotein deficiency type 1 (HDLD1) [MIM:205400]; also known as analphalipoproteinemia or Tangier disease (TGD). HDLD1 is a recessive disorder characterized by the absence of plasma HDL, accumulation of cholesteryl esters, premature coronary artery disease, hepatosplenomegaly, recurrent peripheral neuropathy and progressive muscle wasting and weakness. In HDLD1 patients, ApoA-I fails to associate with HDL probably because of the faulty conversion of pro-ApoA-I molecules into mature chains, either due to a defect in the converting enzyme activity or a specific structural defect in Tangier ApoA-I. DISEASE: Note=A mutation in APOA1 is the cause of amyloid polyneuropathy-nephropathy Iowa type (AMYLIOWA); also known as amyloidosis van Allen type or familial amyloid polyneuropathy type III. AMYLIOWA is a hereditary generalized amyloidosis due to deposition of amyloid mainly constituted by apolipoprotein A1. The clinical picture is dominated by neuropathy in the early stages of the disease and nephropathy late in the course. Death is due in most cases to renal amyloidosis. Severe peptic ulcer disease can occurr in some and hearing loss is frequent. Cataracts is present in several, but vitreous opacities are not observed. DISEASE: Defects in APOA1 are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. SIMILARITY: Belongs to the apolipoprotein A1/A4/E family. WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/APOA1"; WEB RESOURCE: Name=SHMPD; Note=The Singapore human mutation and polymorphism database; URL="http://shmpd.bii.a-star.edu.sg/gene.php?genestart=A&genename=APOA1";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
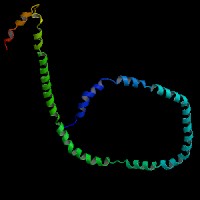
ModBase Predicted Comparative 3D Structure on P02647
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles