Human Gene ATP7B (ENST00000242839.10_8) from GENCODE V47lift37
Description: Copper ion transmembrane transporter involved in the export of copper out of the cells. It is involved in copper homeostasis in the liver, where it ensures the efflux of copper from hepatocytes into the bile in response to copper overload. (from UniProt P35670) Gencode Transcript: ENST00000242839.10_8 Gencode Gene: ENSG00000123191.18_14 Transcript (Including UTRs) Position: hg19 chr13:52,506,805-52,585,586 Size: 78,782 Total Exon Count: 21 Strand: - Coding Region Position: hg19 chr13:52,508,892-52,585,473 Size: 76,582 Coding Exon Count: 21
ID:ATP7B_HUMAN DESCRIPTION: RecName: Full=Copper-transporting ATPase 2; EC=3.6.3.4; AltName: Full=Copper pump 2; AltName: Full=Wilson disease-associated protein; Contains: RecName: Full=WND/140 kDa; FUNCTION: Involved in the export of copper out of the cells, such as the efflux of hepatic copper into the bile. CATALYTIC ACTIVITY: ATP + H(2)O + Cu(2+)(In) = ADP + phosphate + Cu(2+)(Out). SUBUNIT: Monomer. Interacts with COMMD1/MURR1. Interacts with DCTN4, in a copper-dependent manner. Interacts with ATOX1. SUBCELLULAR LOCATION: Golgi apparatus, trans-Golgi network membrane; Multi-pass membrane protein (By similarity). Note=Predominantly found in the trans-Golgi network (TGN). Not redistributed to the plasma membrane in response to elevated copper levels. SUBCELLULAR LOCATION: Isoform 2: Cytoplasm. SUBCELLULAR LOCATION: WND/140 kDa: Mitochondrion. TISSUE SPECIFICITY: Most abundant in liver and kidney and also found in brain. Isoform 2 is expressed in brain but not in liver. The cleaved form WND/140 kDa is found in liver cell lines and other tissues. DOMAIN: Each HMA domain can bind a copper ion, they are tightly packed and closely interact with each other. Wild-type ATP7B can usually be loaded with an average 5.5 copper atoms per molecule. PTM: Isoform 1 may be proteolytically cleaved at the N-terminus to produce the WND/140 kDa form. DISEASE: Defects in ATP7B are the cause of Wilson disease (WD) [MIM:277900]. WD is an autosomal recessive disorder of copper metabolism in which copper cannot be incorporated into ceruloplasmin in liver, and cannot be excreted from the liver into the bile. Copper accumulates in the liver and subsequently in the brain and kidney. The disease is characterized by neurologic manifestations and signs of cirrhosis. SIMILARITY: Belongs to the cation transport ATPase (P-type) (TC 3.A.3) family. Type IB subfamily. SIMILARITY: Contains 6 HMA domains. SEQUENCE CAUTION: Sequence=AAA16173.1; Type=Frameshift; Positions=830; Sequence=AAA79211.1; Type=Frameshift; Positions=456; Sequence=AAA79212.1; Type=Frameshift; Positions=456; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/ATP7B"; WEB RESOURCE: Name=Wilson Disease Mutation Database; URL="http://www.medicalgenetics.med.ualberta.ca/wilson/index.php";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
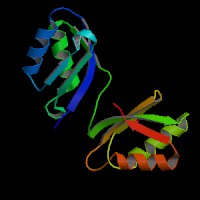
ModBase Predicted Comparative 3D Structure on P35670
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles