ID:CAV3_HUMAN DESCRIPTION: RecName: Full=Caveolin-3; AltName: Full=M-caveolin; FUNCTION: May act as a scaffolding protein within caveolar membranes. Interacts directly with G-protein alpha subunits and can functionally regulate their activity. May also regulate voltage-gated potassium channels. Plays a role in the sarcolemma repair mechanism of both skeletal muscle and cardiomyocytes that permits rapid resealing of membranes disrupted by mechanical stress. SUBUNIT: Homooligomer. Interacts with DLG1 and KCNA5; forms a ternary complex (By similarity). Interacts with TRIM72 (By similarity). Interacts with MUSK; may regulate MUSK signaling (By similarity). Interacts with DAG1 (via its C-terminal); the interaction prevents binding of DAG1 with DMD. Interacts with DYSF. SUBCELLULAR LOCATION: Golgi apparatus membrane; Peripheral membrane protein (By similarity). Cell membrane; Peripheral membrane protein (By similarity). Membrane, caveola; Peripheral membrane protein (By similarity). Note=Potential hairpin-like structure in the membrane. Membrane protein of caveolae (By similarity). TISSUE SPECIFICITY: Expressed predominantly in muscle. PTM: Sumoylation with SUMO3 by PIAS4 may reduce agonist-induced internalization and desensitization of adrenergic receptor ABRD2. DISEASE: Defects in CAV3 are the cause of limb-girdle muscular dystrophy type 1C (LGMD1C) [MIM:607801]. LGMD1C is a myopathy characterized by calf hypertrophy and mild to moderate proximal muscle weakness. LGMD1C inheritance can be autosomal dominant or recessive. DISEASE: Defects in CAV3 are a cause of hyperCKmia (HYPCK) [MIM:123320]. It is a disease characterized by persistent elevated levels of serum creatine kinase without muscle weakness. DISEASE: Defects in CAV3 are a cause of rippling muscle disease (RMD) [MIM:606072]. RMD is a rare disorder characterized by mechanically triggered contractions of skeletal muscle. In RMD, mechanical stimulation leads to electrically silent muscle contractions that spread to neighboring fibers that cause visible ripples to move over the muscle. DISEASE: Defects in CAV3 are a cause of familial hypertrophic cardiomyopathy (CMH) [MIM:192600]; also designated FHC or HCM. Familial hypertrophic cardiomyopathy is a hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. DISEASE: Defects in CAV3 are the cause of long QT syndrome type 9 (LQT9) [MIM:611818]. Long QT syndromes are heart disorders characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to excercise or emotional stress. They can present with a sentinel event of sudden cardiac death in infancy. DISEASE: Defects in CAV3 can be a cause of sudden infant death syndrome (SIDS) [MIM:272120]. SIDS is the sudden death of an infant younger than 1 year that remains unexplained after a thorough case investigation, including performance of a complete autopsy, examination of the death scene, and review of clinical history. Pathophysiologic mechanisms for SIDS may include respiratory dysfunction, cardiac dysrhythmias, cardiorespiratory instability, and inborn errors of metabolism, but definitive pathogenic mechanisms precipitating an infant sudden death remain elusive. Long QT syndromes-associated mutations can be responsible for some SIDS cases. DISEASE: Defects in CAV3 are the cause of myopathy distal Tateyama type (MPDT) [MIM:614321]. A disorder characterized by progressive muscular atrophy and muscle weakness beginning in the hands, the legs, or the feet. Muscle atrophy may be restricted to the small muscles of the hands and feet. SIMILARITY: Belongs to the caveolin family. CAUTION: It is uncertain whether Met-1 or Met-2 is the initiator. SEQUENCE CAUTION: Sequence=AAC14931.1; Type=Erroneous initiation; Note=Translation N-terminally shortened; Sequence=BAF84581.1; Type=Erroneous initiation; Note=Translation N-terminally extended; WEB RESOURCE: Name=CAV3/LGMD1C; Note=Caveolin-3/LGMD-1C page; URL="http://www.dmd.nl/cav3_home.html"; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/CAV3"; WEB RESOURCE: Name=Wikipedia; Note=Caveolin entry; URL="http://en.wikipedia.org/wiki/Caveolin";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
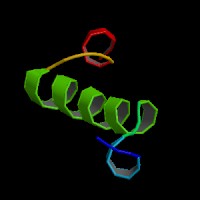
ModBase Predicted Comparative 3D Structure on P56539
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles