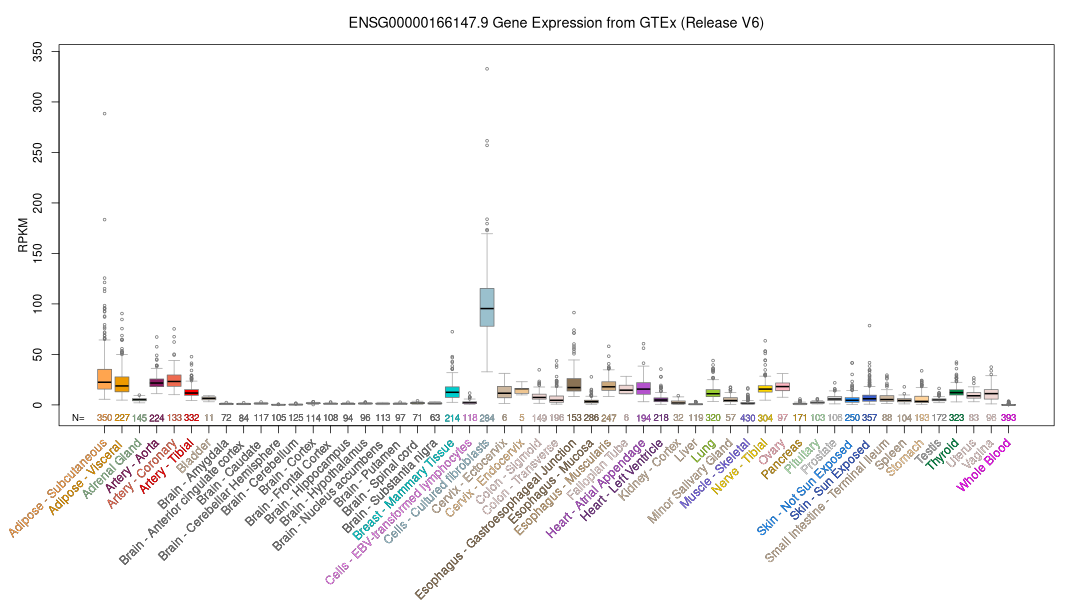
Human Gene FBN1 (ENST00000316623.10_7) from GENCODE V47lift37
Description: [Fibrillin-1]: Structural component of the 10-12 nm diameter microfibrils of the extracellular matrix, which conveys both structural and regulatory properties to load-bearing connective tissues (PubMed:1860873, PubMed:15062093). Fibrillin-1-containing microfibrils provide long-term force bearing structural support (PubMed:27026396). In tissues such as the lung, blood vessels and skin, microfibrils form the periphery of the elastic fiber, acting as a scaffold for the deposition of elastin (PubMed:27026396). In addition, microfibrils can occur as elastin-independent networks in tissues such as the ciliary zonule, tendon, cornea and glomerulus where they provide tensile strength and have anchoring roles (PubMed:27026396). Fibrillin-1 also plays a key role in tissue homeostasis through specific interactions with growth factors, such as the bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs) and latent transforming growth factor-beta-binding proteins (LTBPs), cell-surface integrins and other extracellular matrix protein and proteoglycan components (PubMed:27026396). Regulates osteoblast maturation by controlling TGF- beta bioavailability and calibrating TGF-beta and BMP levels, respectively (By similarity). Negatively regulates osteoclastogenesis by binding and sequestering an osteoclast differentiation and activation factor TNFSF11 (PubMed:24039232). This leads to disruption of TNFSF11-induced Ca(2+) signaling and impairment of TNFSF11-mediated nuclear translocation and activation of transcription factor NFATC1 which regulates genes important for osteoclast differentiation and function (PubMed:24039232). Mediates cell adhesion via its binding to cell surface receptors integrins ITGAV:ITGB3 and ITGA5:ITGB1 (PubMed:12807887, PubMed:17158881). Binds heparin and this interaction has an important role in the assembly of microfibrils (PubMed:11461921). (from UniProt P35555) Gencode Transcript: ENST00000316623.10_7 Gencode Gene: ENSG00000166147.16_10 Transcript (Including UTRs) Position: hg19 chr15:48,700,510-48,937,906 Size: 237,397 Total Exon Count: 66 Strand: - Coding Region Position: hg19 chr15:48,703,187-48,936,966 Size: 233,780 Coding Exon Count: 65
ID:FBN1_HUMAN DESCRIPTION: RecName: Full=Fibrillin-1; Flags: Precursor; FUNCTION: Fibrillins are structural components of 10-12 nm extracellular calcium-binding microfibrils, which occur either in association with elastin or in elastin-free bundles. Fibrillin-1- containing microfibrils provide long-term force bearing structural support. Regulates osteoblast maturation by controlling TGF-beta bioavailability and calibrating TGF-beta and BMP levels, respectively (By similarity). SUBUNIT: Interacts with COL16A1. Interacts with integrin alpha- V/beta-3. Interacts with ADAMTSL4. Interacts with ADAMTS10; this interaction promotes microfibrils assembly. Interacts with THSD4; this interaction promotes fibril formation (By similarity). INTERACTION: O95967:EFEMP2; NbExp=3; IntAct=EBI-2505934, EBI-743414; Q9UBX5:FBLN5; NbExp=3; IntAct=EBI-2505934, EBI-947897; P28300:LOX; NbExp=2; IntAct=EBI-2505934, EBI-3893481; SUBCELLULAR LOCATION: Secreted, extracellular space, extracellular matrix. PTM: Forms intermolecular disulfide bonds either with other fibrillin-1 molecules or with other components of the microfibrils. DISEASE: Defects in FBN1 are a cause of Marfan syndrome (MFS) [MIM:154700]. MFS is an autosomal dominant disorder that affects the skeletal, ocular, and cardiovascular systems. A wide variety of skeletal abnormalities occurs with MFS, including scoliosis, chest wall deformity, tall stature, abnormal joint mobility. Ectopia lentis occurs in up to about 80% of MFS patients and is almost always bilateral. The leading cause of premature death in MFS patients is progressive dilation of the aortic root and ascending aorta, causing aortic incompetence and dissection. Note=The majority of the more than 600 mutations in FBN1 currently known are point mutations, the rest are frameshifts and splice site mutations. Marfan syndrome has been suggested in at least 2 historical figures, Abraham Lincoln and Paganini. DISEASE: Defects in FBN1 are a cause of ectopia lentis, isolated, autosomal dominant (ECTOL1) [MIM:129600]. An ocular abnormality characterized by partial or complete displacement of the lens from its space resulting from defective zonule formation. DISEASE: Defects in FBN1 are the cause of Weill-Marchesani syndrome 2 (WMS2) [MIM:608328]. A rare connective tissue disorder characterized by short stature, brachydactyly, joint stiffness, and eye abnormalities including microspherophakia, ectopia lentis, severe myopia and glaucoma. DISEASE: Defects in FBN1 are a cause of Shprintzen-Goldberg craniosynostosis syndrome (SGS) [MIM:182212]. SGS is a very rare syndrome characterized by a marfanoid habitus, craniosynostosis, characteristic dysmorphic facial features, skeletal and cardiovascular abnormalities, mental retardation, developmental delay and learning disabilities. DISEASE: Defects in FBN1 are a cause of overlap connective tissue disease (OCTD) [MIM:604308]. A heritable disorder of connective tissue characterized by involvement of the mitral valve, aorta, skeleton, and skin. MASS syndrome is closely resembling both the Marfan syndrome and the Barlow syndrome. However, no dislocation of the lenses or aneurysmal changes occur in the aorta, and the mitral valve prolapse is by no means invariable. DISEASE: Defects in FBN1 are a cause of stiff skin syndrome (SSKS) [MIM:184900]. It is a syndrome characterized by hard, thick skin, usually over the entire body, which limits joint mobility and causes flexion contractures. Other occasional findings include lipodystrophy and muscle weakness. DISEASE: Defects in FBN1 are the cause of geleophysic dysplasia type 2 (GPHYSD2) [MIM:614185]. An autosomal dominant disorder characterized by severe short stature, short hands and feet, joint limitations, and skin thickening. Radiologic features include delayed bone age, cone-shaped epiphyses, shortened long tubular bones, and ovoid vertebral bodies. Affected individuals have characteristic facial features including a 'happy' face with full cheeks, shortened nose, hypertelorism, long and flat philtrum, and thin upper lip. Other distinctive features include progressive cardiac valvular thickening often leading to an early death, toe walking, tracheal stenosis, respiratory insufficiency, and lysosomal-like storage vacuoles in various tissues. DISEASE: Defects in FBN1 are the cause of acromicric dysplasia (ACMICD) [MIM:102370]. An autosomal dominant disorder characterized by severe short stature, short hands and feet, joint limitations, and skin thickening. Radiologic features include delayed bone age, cone-shaped epiphyses, shortened long tubular bones, and ovoid vertebral bodies. Affected individuals have distinct facial features, including round face, well-defined eyebrows, long eyelashes, bulbous nose with anteverted nostrils, long and prominent philtrum, and thick lips with a small mouth. Other characteristic features include hoarse voice and pseudomuscular build, and there are distinct skeletal features as well, including an internal notch of the femoral head, internal notch of the second metacarpal, and external notch of the fifth metacarpal. SIMILARITY: Belongs to the fibrillin family. SIMILARITY: Contains 47 EGF-like domains. SIMILARITY: Contains 9 TB (TGF-beta binding) domains. SEQUENCE CAUTION: Sequence=CAA45118.1; Type=Erroneous initiation; Note=Translation N-terminally shortened; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/FBN1";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
ModBase Predicted Comparative 3D Structure on P35555
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Biological Process: GO:0001501 skeletal system development GO:0001656 metanephros development GO:0001822 kidney development GO:0006006 glucose metabolic process GO:0007507 heart development GO:0010469 regulation of receptor activity GO:0010737 protein kinase A signaling GO:0030198 extracellular matrix organization GO:0033627 cell adhesion mediated by integrin GO:0034199 activation of protein kinase A activity GO:0035582 sequestering of BMP in extracellular matrix GO:0035583 sequestering of TGFbeta in extracellular matrix GO:0042593 glucose homeostasis GO:0043010 camera-type eye development GO:0043687 post-translational protein modification GO:0044267 cellular protein metabolic process GO:0045671 negative regulation of osteoclast differentiation GO:0048048 embryonic eye morphogenesis GO:0048050 post-embryonic eye morphogenesis GO:0071560 cellular response to transforming growth factor beta stimulus GO:0090287 regulation of cellular response to growth factor stimulus GO:1990314 cellular response to insulin-like growth factor stimulus GO:2001205 negative regulation of osteoclast development
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles