ID:KIF7_HUMAN DESCRIPTION: RecName: Full=Kinesin-like protein KIF7; FUNCTION: Acts as both a negative and positive regulator of sonic hedgehog (Shh) pathway, acting downstream of SMO. Negatively regulates the pathway by preventing inappropriate activation of the transcriptional activator GLI2 in the absence of ligand. Positively regulates the pathway by preventing the processing of the transcription factor GLI3 into its repressor form. Required for efficient localization of GLI3 to cilia in response to Shh. Affects microtubular dynamics and acts as a ciliary motor. SUBUNIT: Interacts with GLI1, GLI2, GLI3, SMO and SUFU. Interacts with NPHP1. SUBCELLULAR LOCATION: Cell projection, cilium. Note=SMO is required for its accumulation within cilia. Moves from the cilia base to the cilia tip in response to activation of the Shh pathway. TISSUE SPECIFICITY: Embryonic stem cells, melanotic melanoma and Jurkat T-cells. Expressed in heart, lung, liver, kidney, testis, retina, placenta, pancreas, colon, small intestin, prostate and thymus. DISEASE: Note=Ciliary dysfunction leads to a broad spectrum of disorders, collectively termed ciliopathies. The ciliopathy range of diseases includes Meckel-Gruber syndrome, Bardet-Biedl syndrome, Joubert syndrome, and hydrolethalus syndrome among others. Single-locus allelism is insufficient to explain the variable penetrance and expressivity of such disorders, leading to the suggestion that variations across multiple sites of the ciliary proteome influence the clinical outcome. Primary ciliopathy loci can be modulated by pathogenic lesions in other ciliary genes to either exacerbate overall severity or induce specific endophenotypes. KIF7 may be causally associated with diverse ciliopathies, and also acts as a modifier gene across the ciliopathy spectrum. DISEASE: Defects in KIF7 may be a cause of Bardet-Biedl syndrome (BBS) [MIM:209900]. A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease. Note=Heterozygous missense mutations in KIF7 may genetically interact with other BBS genes and contribute to disease manifestation and severity. DISEASE: Defects in KIF7 are the cause of hydrolethalus syndrome type 2 (HLS2) [MIM:614120]. HLS2 is an embryonic lethal disorder characterized by hydrocephaly or anencephaly, postaxial polydactyly of the upper limbs, and pre- or postaxial polydactyly of the lower limbs. Duplication of the hallux is a common finding. DISEASE: Defects in KIF7 are the cause of acrocallosal syndrome (ACLS) [MIM:200990]. ACLS is a syndrome that is characterized by postaxial polydactyly, hallux duplication, macrocephaly and absence of the corpus callosum, usually with severe developmental delay. DISEASE: Defects in KIF7 are the cause of Joubert syndrome type 12 (JBTS12) [MIM:200990]. JBTS12 is a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. DISEASE: Defects in KIF7 may be a cause of Pallister-Hall syndrome (PHS) [MIM:146510]. An autosomal dominant disorder characterized by a wide range of clinical manifestations. Clinical features include hypothalamic hamartoma, pituitary dysfunction, central or postaxial polydactyly, and syndactyly. Malformations are frequent in the viscera, e.g. anal atresia, bifid uvula, congenital heart malformations, pulmonary or renal dysplasia. SIMILARITY: Belongs to the kinesin-like protein family. KIF27 subfamily. SIMILARITY: Contains 1 kinesin-motor domain. SEQUENCE CAUTION: Sequence=AAI04045.1; Type=Erroneous initiation; Note=Translation N-terminally extended; Sequence=AAI12272.1; Type=Erroneous initiation; Note=Translation N-terminally extended; Sequence=AAI12274.1; Type=Erroneous initiation; Note=Translation N-terminally extended; Sequence=AAQ88750.1; Type=Erroneous initiation; Note=Translation N-terminally extended;
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
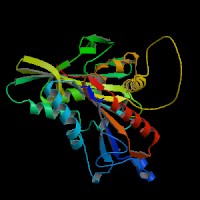
ModBase Predicted Comparative 3D Structure on Q2M1P5
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles