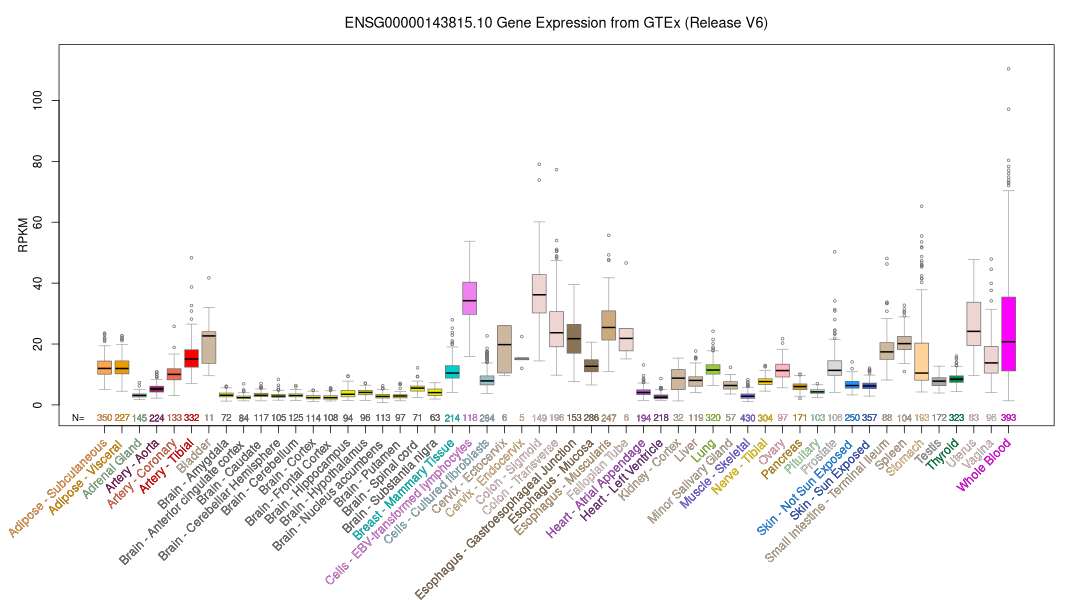
ID:LBR_HUMAN DESCRIPTION: RecName: Full=Lamin-B receptor; AltName: Full=Integral nuclear envelope inner membrane protein; AltName: Full=LMN2R; FUNCTION: Anchors the lamina and the heterochromatin to the inner nuclear membrane. SUBUNIT: Interacts directly with CBX5. Can interact with chromodomain proteins. Interacts directly with DNA. Interaction with DNA is sequence independent with higher affinity for supercoiled and relaxed circular DNA than linear DNA. INTERACTION: Q13185:CBX3; NbExp=4; IntAct=EBI-1055147, EBI-78176; P45973:CBX5; NbExp=4; IntAct=EBI-1055147, EBI-78219; SUBCELLULAR LOCATION: Nucleus inner membrane; Multi-pass membrane protein. DOMAIN: The Tudor domain may not recognize methylation marks, but rather bind unassembled free histone H3 (By similarity). PTM: Phosphorylated by CDK1 in mitosis when the inner nuclear membrane breaks down into vesicles that dissociate from the lamina and the chromatin. It is phosphorylated by different protein kinases in interphase when the membrane is associated with these structures. Phosphorylation of LBR and HP1 proteins may be responsible for some of the alterations in chromatin organization and nuclear structure which occur at various times during the cell cycle. Phosphorylated by SRPK1. In late anaphase LBR is dephosphorylated, probably by PP1 and/or PP2A, allowing reassociation with chromatin. DISEASE: Defects in LBR are a cause of Pelger-Huet anomaly (PHA) [MIM:169400]. PHA is an autosomal dominant inherited abnormality of neutrophils, characterized by reduced nuclear segmentation and an apparently looser chromatin structure. Heterozygotes show hypolobulated neutrophil nuclei with coarse chromatin. Presumed homozygous individuals have ovoid neutrophil nuclei, as well as varying degrees of developmental delay, epilepsy, and skeletal abnormalities. DISEASE: Defects in LBR are the cause of hydrops-ectopic calcification-moth-eaten skeletal dysplasia (HEM) [MIM:215140]; also known as Greenberg skeletal dysplasia. HEM is a rare autosomal recessive chondrodystrophy characterized by early in utero lethality and, therefore, considered to be nonviable. Affected fetuses typically present with fetal hydrops, short- limbed dwarfism, and a marked disorganization of chondro-osseous calcification and may present with polydactyly and additional nonskeletal malformations. DISEASE: Defects in LBR may be a cause of Reynolds syndrome (REYNS) [MIM:613471]. It is a syndrome specifically associating limited cutaneous systemic sclerosis and primary biliray cirrhosis. It is characterized by liver disease, telangiectasia, abrupt onset of digital paleness or cyanosis in response to cold exposure or stress (Raynaud phenomenon), and variable features of scleroderma. The liver disease is characterized by pruritis, jaundice, hepatomegaly, increased serum alkaline phosphatase and positive serum mitochondrial autoantibodies, all consistent with primary biliary cirrhosis. SIMILARITY: Belongs to the ERG4/ERG24 family. SIMILARITY: Contains 1 Tudor domain. SEQUENCE CAUTION: Sequence=BAD92751.1; Type=Erroneous initiation; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/LBR";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
Pfam Domains: PF01222 - Ergosterol biosynthesis ERG4/ERG24 family PF04140 - Isoprenylcysteine carboxyl methyltransferase (ICMT) family PF06966 - Protein of unknown function (DUF1295) PF09465 - Lamin-B receptor of TUDOR domain
ModBase Predicted Comparative 3D Structure on Q14739
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Gene Ontology (GO) Annotations with Structured Vocabulary
Molecular Function: GO:0003677 DNA binding GO:0003723 RNA binding GO:0005515 protein binding GO:0005521 lamin binding GO:0016627 oxidoreductase activity, acting on the CH-CH group of donors GO:0016628 oxidoreductase activity, acting on the CH-CH group of donors, NAD or NADP as acceptor GO:0050613 delta14-sterol reductase activity GO:0070087 chromo shadow domain binding
Biological Process: GO:0006695 cholesterol biosynthetic process GO:0016126 sterol biosynthetic process GO:0055114 oxidation-reduction process
BioCyc Knowledge Library PWY-6074 - zymosterol biosynthesis PWY66-3 - cholesterol biosynthesis II (via 24,25-dihydrolanosterol) PWY66-341 - cholesterol biosynthesis I PWY66-4 - cholesterol biosynthesis III (via desmosterol) PWY66-5 - superpathway of cholesterol biosynthesis
Reactome (by CSHL, EBI, and GO)
Protein Q14739 (Reactome details) participates in the following event(s):
R-HSA-194674 4,4-dimethylcholesta-8(9),14,24-trien-3beta-ol is reduced to 4,4-dimethylcholesta-8(9),24-dien-3beta-ol [LBR] R-HSA-191273 Cholesterol biosynthesis R-HSA-8957322 Metabolism of steroids R-HSA-556833 Metabolism of lipids R-HSA-1430728 Metabolism
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles