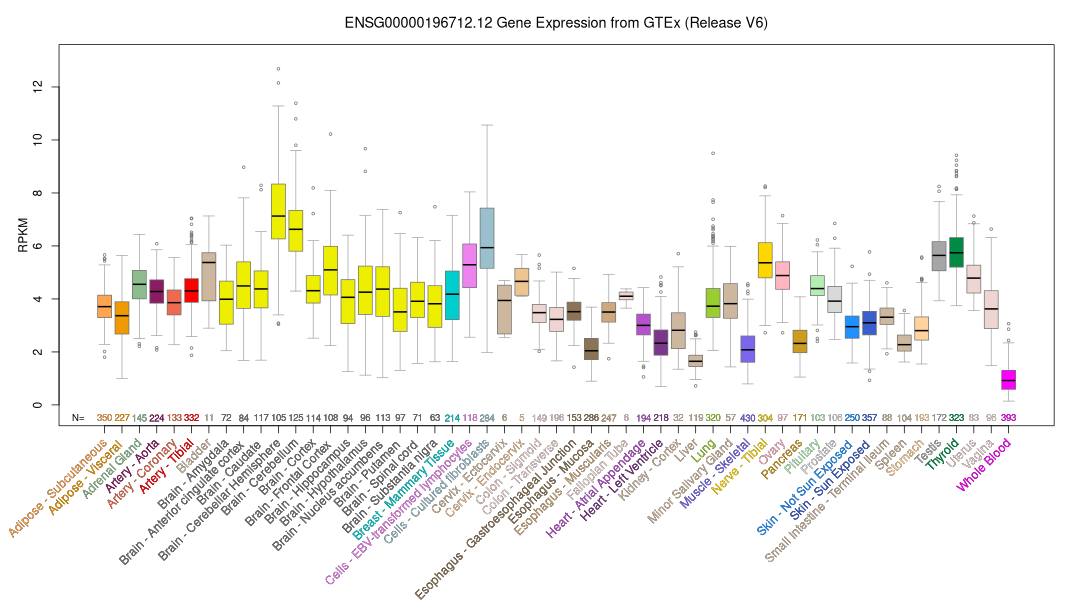
ID:NF1_HUMAN DESCRIPTION: RecName: Full=Neurofibromin; AltName: Full=Neurofibromatosis-related protein NF-1; Contains: RecName: Full=Neurofibromin truncated; FUNCTION: Stimulates the GTPase activity of Ras. NF1 shows greater affinity for Ras GAP, but lower specific activity. May be a regulator of Ras activity. INTERACTION: P05067:APP; NbExp=3; IntAct=EBI-1172917, EBI-77613; P34741:SDC2; NbExp=4; IntAct=EBI-1172917, EBI-1172957; RNA EDITING: Modified_positions=1306; Note=The stop codon (UGA) at position 1306 is created by RNA editing. Various levels of RNA editing occurs in peripheral nerve-sheath tumor samples (PNSTs) from patients with NF1. Preferentially observed in transcripts containing exon 23A. DISEASE: Defects in NF1 are the cause of neurofibromatosis type 1 (NF1) [MIM:162200]; also known as von Recklinghausen syndrome. A disease characterized by patches of skin pigmentation (cafe-au- lait spots), Lisch nodules of the iris, tumors in the peripheral nervous system and fibromatous skin tumors. Individuals with the disorder have increased susceptibility to the development of benign and malignant tumors. DISEASE: Defects in NF1 are a cause of juvenile myelomonocytic leukemia (JMML) [MIM:607785]. JMML is a pediatric myelodysplastic syndrome that constitutes approximately 30% of childhood cases of myelodysplastic syndrome (MDS) and 2% of leukemia. Germline mutations of NF1 account for the association of JMML with type 1 neurofibromatosis (NF1). DISEASE: Defects in NF1 are the cause of Watson syndrome (WS) [MIM:193520]. WS is characterized by the presence of pulmonary stenosis, cafe-au-lait spots, and mental retardation. WS is considered as an atypical form of NF1. DISEASE: Defects in NF1 are a cause of familial spinal neurofibromatosis (FSNF) [MIM:162210]. Familial spinal NF is considered to be an alternative form of neurofibromatosis, showing multiple spinal tumors. DISEASE: Defects in NF1 are a cause of neurofibromatosis-Noonan syndrome (NFNS) [MIM:601321]. NFNS is characterized by manifestations of both NF1 and Noonan syndrome (NS). NS is a disorder characterized by dysmorphic facial features, short stature, hypertelorism, cardiac anomalies, deafness, motor delay, and a bleeding diathesis. DISEASE: Defects in NF1 may be a cause of colorectal cancer (CRC) [MIM:114500]. SIMILARITY: Contains 1 CRAL-TRIO domain. SIMILARITY: Contains 1 Ras-GAP domain. CAUTION: Was originally (PubMed:8807336) thought to be associated with LEOPARD (LS), an autosomal dominant syndrome. SEQUENCE CAUTION: Sequence=AAA59923.1; Type=Erroneous initiation; WEB RESOURCE: Name=NF1 Genetic Mutation Analysis Consortium; URL="http://www.upmc.edu/Neurofibro/NNFFconsortium.htm"; WEB RESOURCE: Name=Atlas of Genetics and Cytogenetics in Oncology and Haematology; URL="http://atlasgeneticsoncology.org/Genes/NF1ID134.html"; WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/NF1"; WEB RESOURCE: Name=NIEHS-SNPs; URL="http://egp.gs.washington.edu/data/nf1/"; WEB RESOURCE: Name=Mendelian genes neurofibromin 1 (NF1); Note=Leiden Open Variation Database (LOVD); URL="http://www.lovd.nl/NF1";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
ModBase Predicted Comparative 3D Structure on P21359
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles