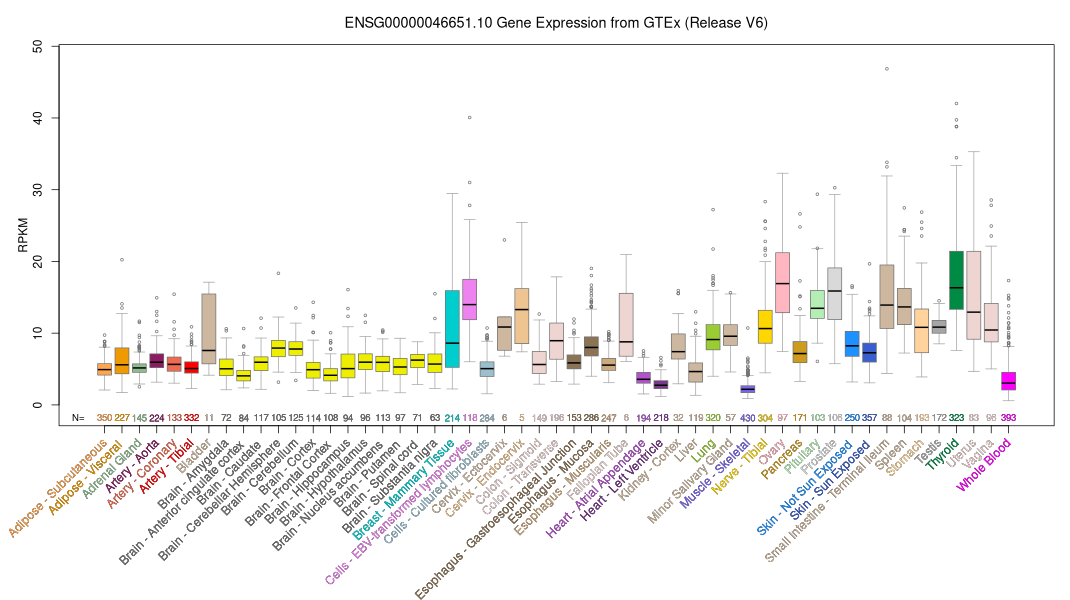
ID:OFD1_HUMAN DESCRIPTION: RecName: Full=Oral-facial-digital syndrome 1 protein; AltName: Full=Protein 71-7A; FUNCTION: Component of the centrioles controlling mother and daughter centrioles length. Recruits to the centriole IFT88 and centriole distal appendage-specific proteins including CEP164. Involved in the biogenesis of the cilium, a centriole-associated function. The cilium is a cell surface projection found in many vertebrate cells required to transduce signals important for development and tissue homeostasis. Plays an important role in development by regulating Wnt signaling and the specification of the left-right axis (By similarity). SUBUNIT: Homooligomer. Interacts with LCA5. Interacts with RUVBL1; the interaction is direct and may mediate interaction with the NuA4 histone acetyltransferase complex. Interacts with SDCCAG8; the interaction is direct. INTERACTION: P53350:PLK1; NbExp=2; IntAct=EBI-716327, EBI-476768; SUBCELLULAR LOCATION: Cytoplasm, cytoskeleton, centrosome, centriole. Cytoplasm, cytoskeleton, cilium basal body. Nucleus. Note=Localizes to centriole distal ends (By similarity). TISSUE SPECIFICITY: Widely expressed. Expressed in 9 and 14 weeks old embryos in metanephric mesenchyme, oral mucosa, lung, heart, nasal and cranial cartilage, and brain. Expressed in metanephros, brain, tongue, and limb. DISEASE: Defects in OFD1 are the cause of oral-facial-digital syndrome type 1 (OFD1) [MIM:311200]. OFD1 is a X-linked dominant condition with lethality in males. The syndrome is characterized by clefts of the jaw and tongue in the area of the lateral incisors and canines. Other features are malformations of the face and skull, malformation of the hands (specifically syndactyly, clinodactyly, brachydactyly and occasionally postaxial polydactyly) and mental retardation. OFD1 also causes polycystic kidney disease. DISEASE: Defects in OFD1 are associated with Simpson-Golabi-Behmel syndrome type 2 (SGBS2) [MIM:300209]. SGBS2 is a severe variant of Simpson-Golabi-Behmel syndrome, a condition characterized by pre- and postnatal overgrowth (gigantism), facial dysmorphism and a variety of inconstant visceral and skeletal malformations. DISEASE: Defects in OFD1 are the cause of Joubert syndrome type 10 (JBTS10) [MIM:300804]. A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. SIMILARITY: Belongs to the OFD1 family. SIMILARITY: Contains 1 LisH domain. WEB RESOURCE: Name=GeneReviews; URL="http://www.ncbi.nlm.nih.gov/sites/GeneTests/lab/gene/OFD1";
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
ModBase Predicted Comparative 3D Structure on O75665
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
AK290354 - Homo sapiens cDNA FLJ78705 complete cds, highly similar to Homo sapiens oral-facial-digital syndrome 1 (OFD1), mRNA. AK289677 - Homo sapiens cDNA FLJ75575 complete cds, highly similar to Oral-facial-digital syndrome 1 protein (Protein 71-7A). BC012324 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:4555096), with apparent retained intron. BC030787 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:5267734), with apparent retained intron. BC062432 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:6495872), partial cds. Y15164 - Homo sapiens mRNA for protein encoded by cxorf5 (71-7A) gene. Y16355 - Homo sapiens mRNA for protein encoded by cxorf5 (71-7A) gene, alternatively spliced form. BC042830 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:5302058), complete cds. AK225847 - Homo sapiens mRNA for Splice isoform 2 of O75665 variant, clone: FCC126B11. BC052809 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:6151125), partial cds. BC096344 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone MGC:117038 IMAGE:40008704), complete cds. BC096345 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:40008707), complete cds. BC099658 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:40008705), with apparent retained intron. BC099659 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:40008706), with apparent retained intron. BC092448 - Homo sapiens oral-facial-digital syndrome 1, mRNA (cDNA clone IMAGE:30350282), containing frame-shift errors. GU727634 - Homo sapiens epididymis tissue sperm binding protein Li 5a mRNA, complete cds. JD301792 - Sequence 282816 from Patent EP1572962. AK311067 - Homo sapiens cDNA, FLJ18109. AK297104 - Homo sapiens cDNA FLJ52326 complete cds, highly similar to Oral-facial-digital syndrome 1 protein. JD146815 - Sequence 127839 from Patent EP1572962. JD233378 - Sequence 214402 from Patent EP1572962. JD346264 - Sequence 327288 from Patent EP1572962.
Biochemical and Signaling Pathways
Reactome (by CSHL, EBI, and GO)
Protein O75665 (Reactome details) participates in the following event(s):
R-HSA-380272 Plk1-mediated phosphorylation of Nlp R-HSA-380283 Recruitment of additional gamma tubulin/ gamma TuRC to the centrosome R-HSA-380294 Loss of C-Nap-1 from centrosomes R-HSA-380311 Recruitment of Plk1 to centrosomes R-HSA-380455 Recruitment of CDK11p58 to the centrosomes R-HSA-380303 Dissociation of Phospho-Nlp from the centrosome R-HSA-5626220 C2CD3 binds the mother centriole R-HSA-380508 Translocation of NuMA to the centrosomes R-HSA-2574845 AJUBA binds centrosome-associated AURKA R-HSA-8853405 TPX2 binds AURKA at centrosomes R-HSA-3000319 BORA binds PLK1 and AURKA R-HSA-2574840 AJUBA facilitates AURKA autophosphorylation R-HSA-3000310 AURKA phosphorylates PLK1 R-HSA-5626223 C2CD3 and OFD1 recruit 5 distal appendage proteins to the centriole R-HSA-5626681 Recruitment of transition zone proteins R-HSA-5626227 CP110 and CEP97 dissociate from the centriole R-HSA-380316 Association of NuMA with microtubules R-HSA-8853419 TPX2 promotes AURKA autophosphorylation R-HSA-5626228 The distal appendage proteins recruit TTBK2 R-HSA-5638009 CEP164 recruits RAB3IP-carrying Golgi-derived vesicles to the basal body R-HSA-5626699 MARK4 binds ODF2 in the centriole R-HSA-5617816 RAB3IP stimulates nucleotide exchange on RAB8A R-HSA-380259 Loss of Nlp from mitotic centrosomes R-HSA-380270 Recruitment of mitotic centrosome proteins and complexes R-HSA-380284 Loss of proteins required for interphase microtubule organization from the centrosome R-HSA-5620912 Anchoring of the basal body to the plasma membrane R-HSA-380320 Recruitment of NuMA to mitotic centrosomes R-HSA-2565942 Regulation of PLK1 Activity at G2/M Transition R-HSA-8854518 AURKA Activation by TPX2 R-HSA-380287 Centrosome maturation R-HSA-5617833 Cilium Assembly R-HSA-68877 Mitotic Prometaphase R-HSA-69275 G2/M Transition R-HSA-1852241 Organelle biogenesis and maintenance R-HSA-68886 M Phase R-HSA-453274 Mitotic G2-G2/M phases R-HSA-69278 Cell Cycle (Mitotic) R-HSA-1640170 Cell Cycle
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles