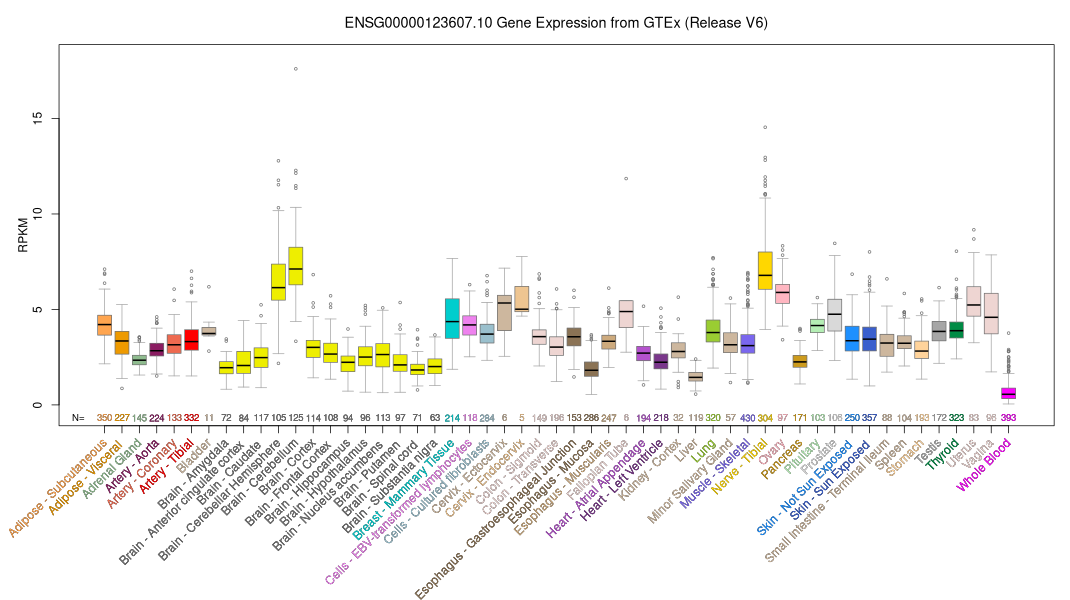
ID:TT21B_HUMAN DESCRIPTION: RecName: Full=Tetratricopeptide repeat protein 21B; Short=TPR repeat protein 21B; FUNCTION: May negatively modulate SHH signal transduction and may play a role in retrograde intraflagellar transport in cilia (By similarity). SUBCELLULAR LOCATION: Cytoplasm, cytoskeleton, cilium axoneme (By similarity). DISEASE: Note=Ciliary dysfunction leads to a broad spectrum of disorders, collectively termed ciliopathies. Overlapping clinical features include retinal degeneration, renal cystic disease, skeletal abnormalities, fibrosis of various organ, and a complex range of anatomical and functional defects of the central and peripheral nervous system. The ciliopathy range of diseases includes Meckel-Gruber syndrome, Bardet-Biedl syndrome, Joubert syndrome, nephronophtisis, Senior-Loken syndrome, and Jeune asphyxiating thoracic dystrophy among others. TTC21B is causally associated with diverse ciliopathies, and also acts as a modifier gene across the ciliopathy spectrum. TTC21B mutations interact in trans with mutations in other ciliopathy-causing genes and contribute to disease manifestation and severity. DISEASE: Defects in TTC21B are the cause of nephronophthisis type 12 (NPHP12) [MIM:613820]. NPHP12 is an autosomal recessive disorder resulting in end-stage renal disease. It is a progressive tubulo-interstitial kidney disorder histologically characterized by modifications of the tubules with thickening of the basement membrane, interstitial fibrosis and, in the advanced stages, medullary cysts. Some patients manifest extra-renal features including retinal, skeletal and central nervous system defects. DISEASE: Defects in TTC21B are the cause of asphyxiating thoracic dystrophy type 4 (ATD4) [MIM:613819]. ATD4 is an autosomal recessive chondrodysplasia characterized by a severely constricted thoracic cage, short-limbed short stature, and polydactyly. It often leads to death in infancy because of respiratory insufficiency. Retinal degeneration, cystic renal disease and hepatic disease can be present in affected individuals who survive early childhood. DISEASE: Defects in TTC21B may be a cause of Bardet-Biedl syndrome (BBS) [MIM:209900]. A syndrome characterized by usually severe pigmentary retinopathy, early-onset obesity, polydactyly, hypogenitalism, renal malformation and mental retardation. Secondary features include diabetes mellitus, hypertension and congenital heart disease. Bardet-Biedl syndrome inheritance is autosomal recessive, but three mutated alleles (two at one locus, and a third at a second locus) may be required for clinical manifestation of some forms of the disease. DISEASE: Defects in TTC21B may be a cause of Joubert syndrome (JBTS) [MIM:213300]. A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. SIMILARITY: Belongs to the TTC21 family. SIMILARITY: Contains 19 TPR repeats. SEQUENCE CAUTION: Sequence=AAY14750.1; Type=Erroneous gene model prediction; Sequence=BAB13836.1; Type=Erroneous initiation; Note=Translation N-terminally extended;
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
ModBase Predicted Comparative 3D Structure on Q7Z4L5
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.
Gene Ontology (GO) Annotations with Structured Vocabulary
Biological Process: GO:0006357 regulation of transcription from RNA polymerase II promoter GO:0007224 smoothened signaling pathway GO:0008589 regulation of smoothened signaling pathway GO:0021591 ventricular system development GO:0021798 forebrain dorsal/ventral pattern formation GO:0035721 intraciliary retrograde transport GO:0035735 intraciliary transport involved in cilium assembly GO:0061512 protein localization to cilium
 Sequence and Links to Tools and Databases
Sequence and Links to Tools and Databases  Common Gene Haplotype Alleles
Common Gene Haplotype Alleles