Description
These tracks provide heatmaps of chromatin folding data from in situ Hi-C experiments on
the seven cell lines (Rao et al., 2014). Two other cell lines were
also part of this project, but are not included in this track: CH12-LX (mouse B-lymphoblasts)
and HeLa (the Henrietta Lacks tumor cell line). Below are the seven types of cells sequenced
with a short description:
| GM12878 |
B-Lymphocyte Cells |
| HMEC |
Mammary Epithelial Cells |
| HUVEC |
Umbilical Endothelial Cells |
| IMR90 |
Fetal Lung Cells |
| K562 |
Immortalised Leukemia Cells |
| KBM7 |
Immortalised Leukemia Cells |
| NHEK |
Epidermal Keratinocyte Cells |
The data indicate how many interactions were detected between regions of the genome. A high score
between two regions suggests that they are probably in close proximity in 3D space within the
nucleus of a cell. In the track display, this is shown by a more intense color in the heatmap.
Display Conventions
This is a composite track with data from seven cell lines. Individual subtrack settings can be
adjusted by clicking the wrench next to the subtrack name, and all subtracks can be configured
simultaneously using the track controls at the top of the page. Note that some controls
(specifically, resolution and normalization options) are only available in the subtrack-specific
configuration. The proximity data in these tracks are displayed as heatmaps, with high scores (and
more intense colors) corresponding to closer proximity.
Draw modes
There are three display methods available for Hi-C heatmaps: square, triangle, and arc.
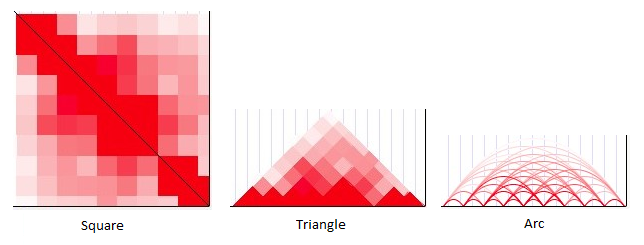
Square mode provides a traditional Hi-C display in which chromosome positions are mapped along the
top-left-to-bottom-right diagonal, and interaction values are plotted on both sides of that diagonal
to form a square. The upper-left corner of the square corresponds to the left-most position of the
window in view, while the bottom-right corner corresponds to the right-most position of the window.
The color shade at any point within the square shows the proximity score for two genomic regions:
the region where a vertical line drawn from that point intersects with the diagonal, and the region
where a horizontal line from that point intersects with the diagonal. A point directly on the
diagonal shows the score for how proximal a region is to itself (scores on the diagonal are usually
quite high unless no data are available). A point at the extreme bottom left of the square shows the
score for how proximal the left-most position within the window is to the right-most position within
the window.
In triangle mode, the display is quite similar to square except that only the top half of the square
is drawn (eliminating the redundancy), and the image is rotated so that the diagonal of the square
now lies on the horizontal axis. This display consumes less vertical space in the image, although it
may be more difficult to ascertain exactly which positions correspond to a point within the
triangle.
In arc mode, simple arcs are drawn between the centers of interacting regions. The color of each arc
corresponds to the proximity score. Self-interactions are not displayed.
Score normalization settings
Score values for this type of display correspond to how close two genomic regions are in 3D space. A
high score indicates more links were formed between them in the experiment, which suggests that the
regions are near to each other. A low score suggests that the regions are farther apart. High scores
are displayed with a more intense color value; low scores are displayed in paler shades.
There are four score values available in this display: NONE, VC, VC_SQRT, and KR. NONE provides raw,
un-normalized counts for the number of interactions between regions. VC, or Vanilla Coverage,
normalization (Lieberman-Aiden et al., 2009) and the VC_SQRT variant normalize these count
values based on the overall count values for each of the two interacting regions. Knight-Ruiz, or
KR, matrix balancing (Knight and Ruiz, 2013) provides an alternative normalization method where the
row and column sums of the contact matrix equal 1.
Color intensity in the heatmap goes up to indicate higher scores, but eventually saturates at a
maximum beyond which all scores share the same color intensity. The value of this maximum score for
saturation can be set manually by un-checking the "Auto-scale" box. When the
"Auto-scale" box is checked, it automatically sets the saturation maximum to be double
(2x) the median score in the current display window.
Resolution settings
The resolution for each track is measured in base pairs and represents the size of the bins into
which proximity data are gathered. The list of available resolutions ranges from 1kb to 10Mb. There
is also an "Auto" setting, which attempts to use the coarsest resolution that still
displays at least 500 bins in the current window.
Methods
The protocol described in this paper, in situ Hi-C, is a refinement of an earlier method
originally called Hi-C and now referred to as dilution Hi-C. Both methods involve cross-linking DNA
with formaldehyde, cleaving it with a restriction enzyme, forming local bonds between the cleaved
DNA ends, and sequencing the resulting junctions. The primary refinement for in situ Hi-C is that it
keeps cell nuclei intact during cross-linking, which reduces the number of spurious contacts in the
resulting contact matrix. The protocol also takes less time (3 days instead of 7) and can make use
of higher-resolution restriction enzymes.
The cell lines in this paper were processed using the in situ Hi-C protocol to produce contact
matrices in the .hic format. We downloaded a subset of those files from the GEO repository at
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE63525. The files used for
this track are the "combined.hic" files, which combine the results from multiple
experimental replicates without imposing a cutoff on the data values. The files are parsed for
display using the Straw library from
the Aiden lab at Baylor College of Medicine.
Data Access
The data for this track can be explored interactively with the Table Browser in the
interact format. Direct access to the
raw data files in .hic format can be obtained from GEO at the URL provided in the Methods section or
from our own download server.
The following files for this track can be found in the
/gbdb/hg19/hic/
subdirectory:
GSE63525_GM12878_insitu_primary+replicate_combined.hic, GSE63525_HUVEC_combined.hic,
GSE63525_K562_combined.hic, GSE63525_NHEK_combined.hic, GSE63525_HMEC_combined.hic,
GSE63525_IMR90_combined.hic, and GSE63525_KBM7_combined.hic. Details on working with .hic files
can be found at https://www.aidenlab.org/documentation.html.
References
Knight P, Ruiz D.
A fast algorithm for matrix balancing.
IMA J Numer Anal. 2013 Jul;33(3):1029-1047.
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR,
Sabo PJ, Dorschner MO et al.
Comprehensive mapping of long-range interactions reveals folding principles of the human genome.
Science. 2009 Oct 9;326(5950):289-93.
PMID: 19815776; PMC: PMC2858594
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD,
Lander ES et al.
A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping.
Cell. 2014 Dec 18;159(7):1665-80.
PMID: 25497547; PMC: PMC5635824
|
|