- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
Docker Help Page
Contents
What is Docker?
How to Install Docker Desktop?
Using Docker Desktop for UCSC Genome Browser
Create a Docker Volume for Data Persistence
Updating the Latest UCSC Genome Browser Version
Customize a UCSC Genome Browser Docker Container
What is Docker?
Docker is a platform for developing, testing and running applications. Docker can be used to run genomics tools and manage software such as the UCSC Genome Browser. Docker offers consistency across different computers and environments by packaging everything needed including specific software versions and configurations into a self-contained unit called a container.
Container
A container is software that packages up code and all its dependencies to run an application quickly and reliably from one computing environment to another. A container is isolated from other containers and a Docker container can be run on a developer's local laptop, virtual machines, on cloud providers, or other combinations of environments. A genomics analysis pipeline or entire analysis environment can be packaged to a local computer into a Docker container and moved to a cluster or a cloud server. See also: Use containers to Build, Share and Run applications
Image
A Dockerfile is a text file that provides instructions to build an image. The Dockerfile is written in Dockerfile syntax. A docker image is a read-only template with instructions and everything needed to run an application for the container. See also: Overview of the get started guide
How to Install Docker Desktop?
Windows
- Go to the Install Docker Desktop on Windows page
- Check system requirements
- Install Docker interactively or from the command line
macOS
- Go to the Install and run Docker Desktop on Mac page
- Check system requirements
- Install Docker interactively or from the command line
Linux
- Go to the Install Docker Desktop on Linux page and select Linux distribution
- Check system requirements
- Follow Generic installation steps
Using Docker Desktop for UCSC Genome Browser
Start Docker Desktop after installation is complete:
- Windows: start Docker Desktop from the Start menu
- macOS: start Docker Desktop from the Applications folder
- Linux: start the Docker service by running the following command on the terminal:
sudo systemctl start docker
Obtaining a UCSC Genome Browser Dockerfile
The UCSC Genome Browser dockerfile can be obtained from the UCSC Genome Browser Github by using the wget command:
wget https://raw.githubusercontent.com/ucscGenomeBrowser/kent/master/src/product/installer/docker/DockerfileCreating a Image
Once the dockerfile has been downloaded, running the docker build with the 't' option allows the naming and the optional tag (format: "name:tag") of the image. The image can be created by running the following command in the same directory where the dockerfile is located:
docker build . -t user_name/ucsc_genomebrowser_imageCreating a Container
After the image has been created, running the docker run command and the image with the -d option allows the container to be run in the background, whereas the default runs the container in the foreground. The -p option publishes a container's port(s) to the host. The following command maps port 8080 on the host machine to port 80 in the container and names the container using the -name option:
docker run -d --name ucsc_genomebrowser_container -p 8080:80 user_name/ucsc_genomebrowser_imageAccessing the running container via http://localhost:8080
Running the following command will list the running container:
docker container lsRunning the following command stops the running container::
docker stop <container_name_or_id>Running the following command removes the existing container::
docker rm <container_name_or_id>Using Docker Desktop to Create a Container
The Docker Desktop user interface can be used to run the container by going to the images tab and clicking the run button under Actions:
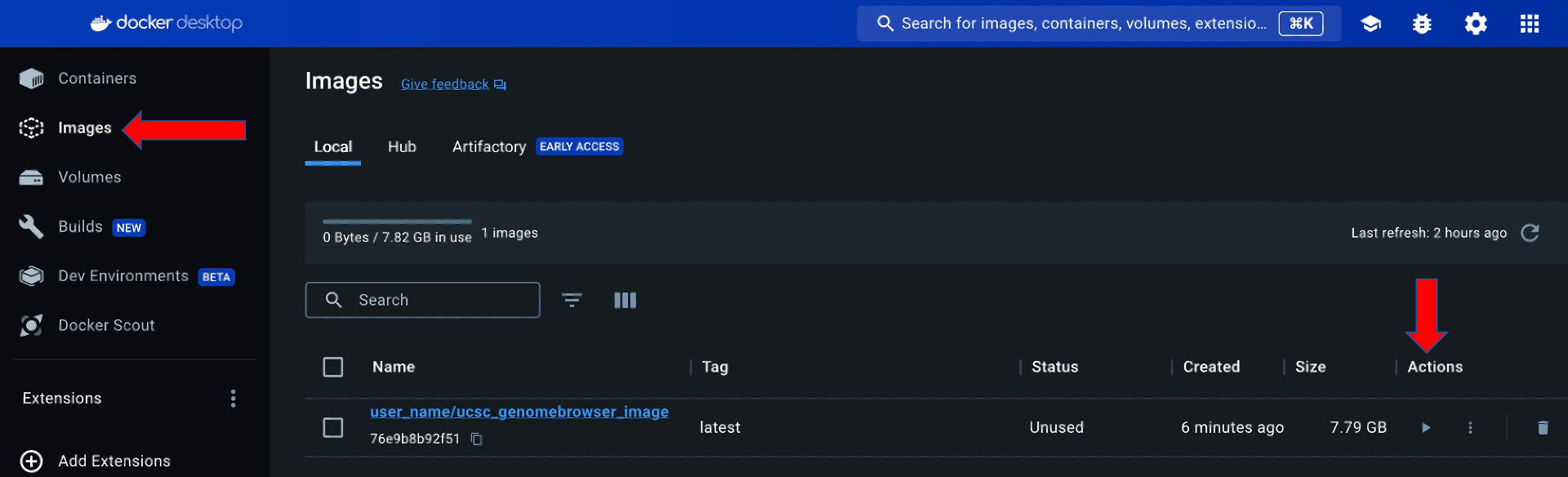
Click Optional settings in the "Run a new container" pop-up window:
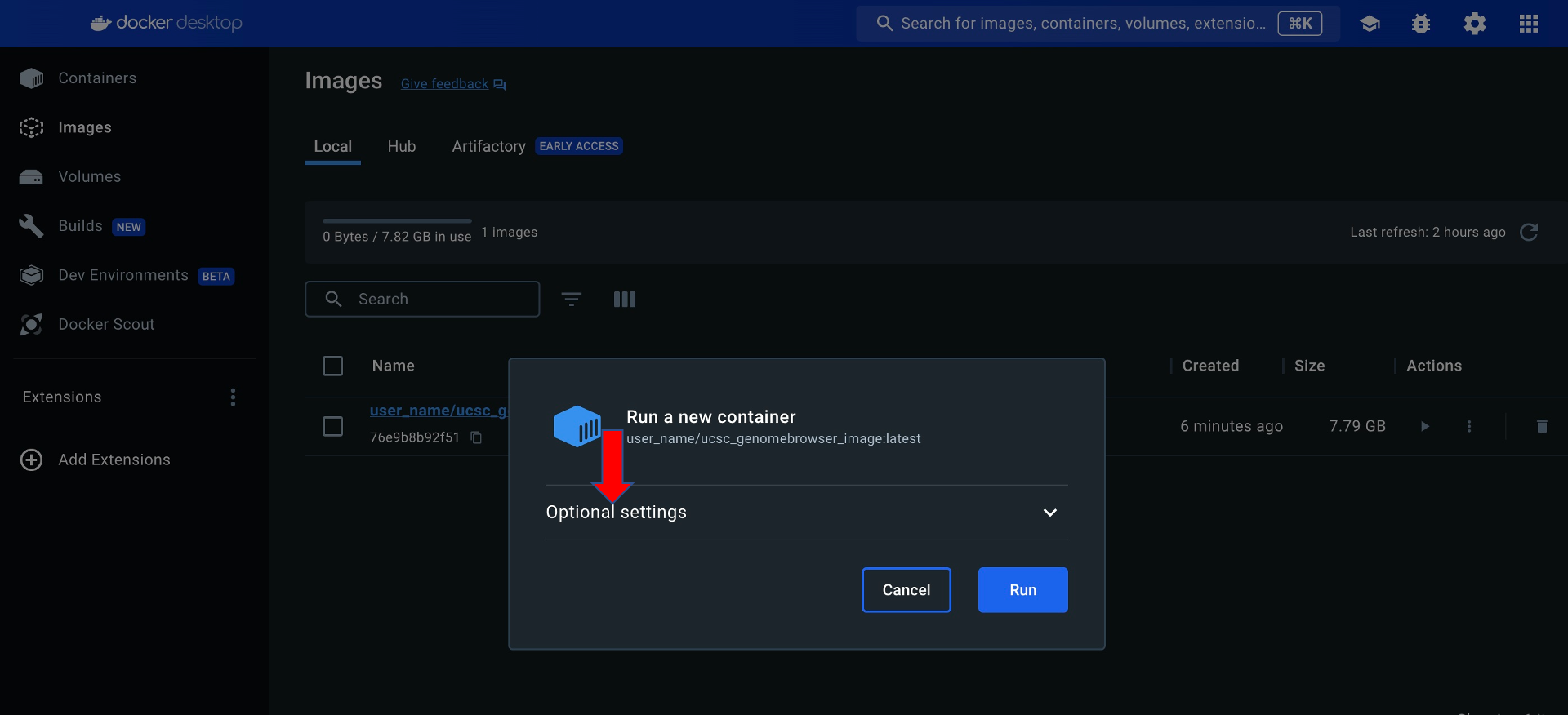
Enter a Container name and a Host port in the "Run a new container" popup window:
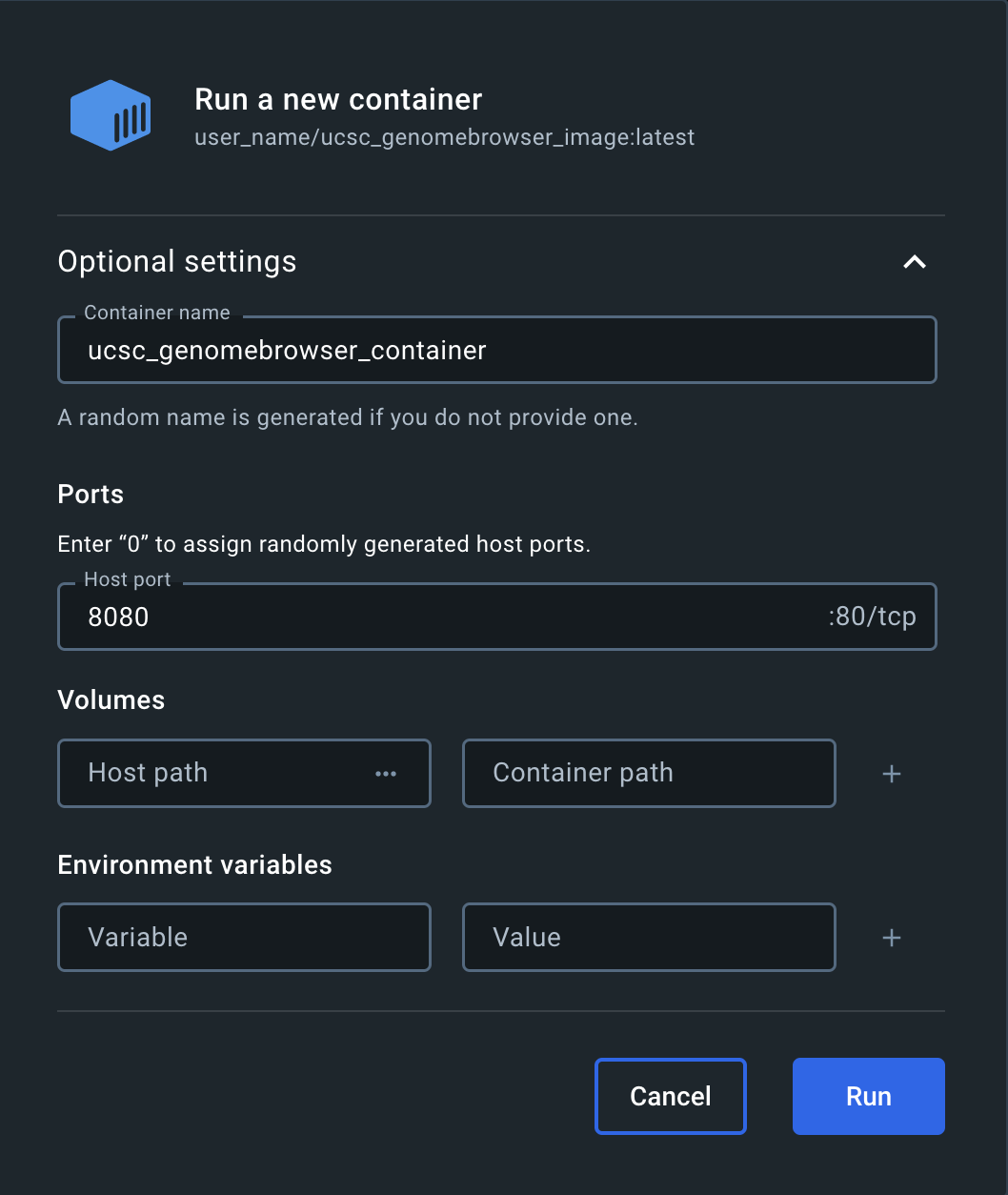
Click the link with the Host port to go to the running container via localhost:
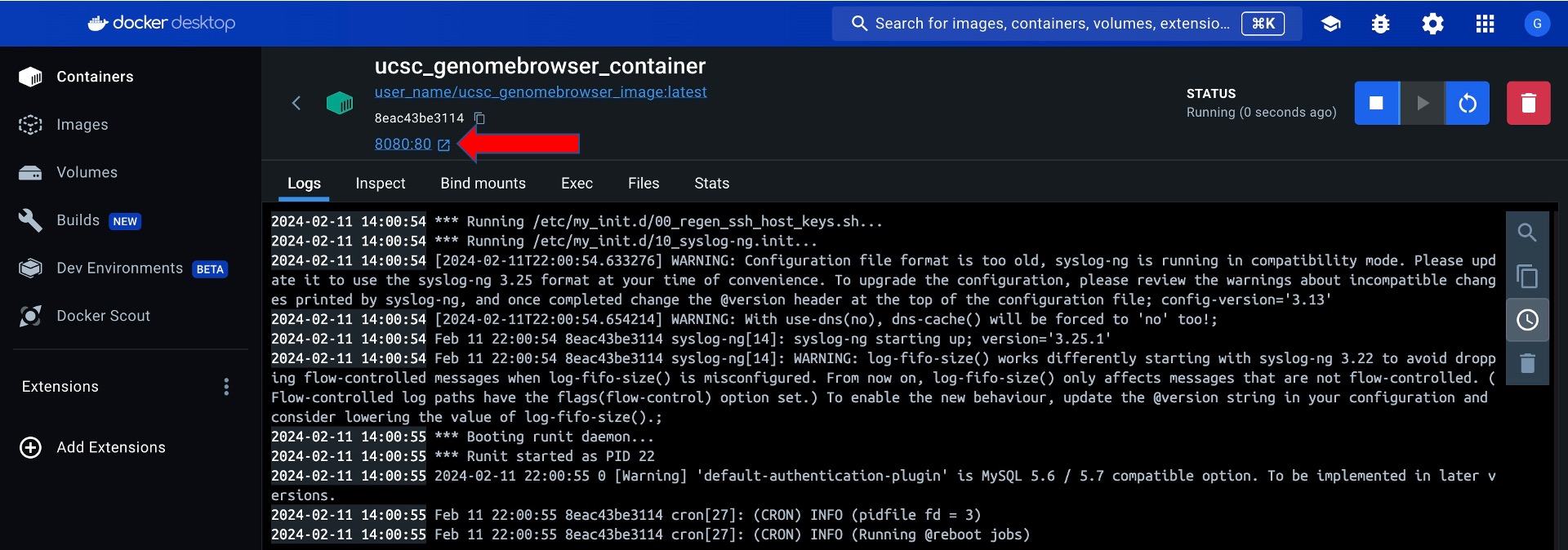
Create a Docker Volume for Data Persistence
A Docker volume allows the data to be persistent (long-lasting) after the container restarts and mount to a host directory or another container's data volume into the UCSC Genome Browser container.
The following command creates a new volume named ucsc_genomebrowser_volume that containers can consume and store data in:
docker volume create ucsc_genomebrowser_volumeAfter creating the volume named ucsc_genomebrowser_volume, running the docker run command starts the UCSC Genome Browser container using the user_name/ucsc_genomebrowser_image image and the -v option to mount the volume created in the previous step.
docker run -d --name ucsc_genomebrowser_volume -p 8080:80 -v ucsc_genomebrowser_volume:/data user_name/ucsc_genomebrowser_imageFiles can be copied into the Docker volume or a bind mount can be used to link a host directory containing data to the /data directory inside the container. The following command copies a file to the data directory inside the container:
docker cp file.txt ucsc_genomebrowser_volume:/dataRunning the execute command will list the file inside the running container:
docker exec ucsc_genomebrowser_volume ls dataUpdating the Latest UCSC Genome Browser Software
Access the Docker Container's Shell
Updating the latest UCSC Genome Browser version will require access to the Docker container running shell (command-line interface) of the UCSC Genome Browser. The execute command can be run inside a running Docker container with the -it options. The -i or --interactive option allows interaction with the command being executed and keeps STDIN open even if not attached. This will allow input to be provided for the command. The -t or --tty option allocates a pseudo-TTY and allows for a more interactive experience. The following example shows how to run exec command and the -it options:
docker exec -it <container_name_or_id> /bin/bashUpdate the Genome Browser Software
Running the following command updates the Genome Browser software:
bash root/browserSetup.sh cgiUpdateCustomize a UCSC Genome Browser Docker Container
Editing hg.conf
The hg.conf file is a file that has information on how to connect to MariaDB, the location of the other directories and various other settings.
The hg.conf file can be edited by running the execute command inside a running Docker container with the -it options. The -i or --interactive option allows interaction with the command being executed and keeps STDIN open even if not attached. This will allow input to be provided for the command. The -t or --tty option allocates a pseudo-TTY and allows for a more interactive experience. Any common text editors such as vi, nano, and vim can be used with the execute command and the -it options. The following example shows how to edit the hg.conf file using vi:
docker exec -it <container_name_or_id> vi /usr/local/apache/cgi-bin/hg.confChanging the Default Genome Browser Options
Track settings such as fonts, text size, default tracks, attached hubs, and the default region can be customized and set as the default settings. These settings will appear every time the UCSC Genome Browser graphic display is opened and will also appear after a reset of all user settings. This can be useful when working with a different assembly than hg38, having track hubs automatically attached, or changing the visibility of tracks.
- The first step is to create a Session containing all desired display options, hubs, tracks, and default region in the UCSC Genome Browser Docker container. Open the UCSC Genome Browser Docker container shell. See the Access the Docker Container's Shell section of this page.
- Create a new file, 'defaultCart.sql', to make a new MySQL table. Add the following to the defaultCart.sql file:
#The default cart CREATE TABLE defaultCart ( contents longblob not null # cart contents ); - Drop the existing defaultCart table by running the following query:
mysql hgcentral -Ne "DROP TABLE defaultCart"
- Load the defaultCart.sql file as a table by running the following query:
mysql hgcentral < defaultCart.sql
- Insert the session to the default cart table by using the user name and the session name,
which was the session saved in the earlier step, and run the following query (add userName):
mysql hgcentral -Ne "insert into defaultCart select contents from namedSessionDb where sessionName='nameOfSession' and userName='nameOfUser'"