- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
Chain Format
The chain format describes a pairwise alignment that allow gaps in both sequences simultaneously. Each set of chain alignments starts with a header line, contains one or more alignment data lines, and terminates with a blank line. The format is deliberately quite dense.
Example:
chain 4900 chrY 58368225 + 25985403 25985638 chr5 151006098 - 43257292 43257528 1
9 1 0
10 0 5
61 4 0
16 0 4
42 3 0
16 0 8
14 1 0
3 7 0
48
chain 4900 chrY 58368225 + 25985406 25985566 chr5 151006098 - 43549808 43549970 2
16 0 2
60 4 0
10 0 4
70Header Lines
chain score tName tSize tStrand tStart tEnd qName qSize qStrand qStart qEnd id
The initial header line starts with the keyword chain, followed by
11 required attribute values, and ends with a blank line. The attributes include:
-
score-- chain score -
tName-- chromosome (reference/target sequence) -
tSize-- chromosome size (reference/target sequence) -
tStrand-- strand (reference/target sequence) -
tStart-- alignment start position (reference/target sequence) -
tEnd-- alignment end position (reference/target sequence) -
qName-- chromosome (query sequence) -
qSize-- chromosome size (query sequence) -
qStrand-- strand (query sequence) -
qStart-- alignment start position (query sequence) -
qEnd-- alignment end position (query sequence) -
id-- chain ID
The alignment start and end positions are represented as zero-based half-open intervals. For example, the first 100 bases of a sequence would be represented with start position = 0 and end position = 100, and the next 100 bases would be represented as start position = 100 and end position = 200. When the strand value is "-", position coordinates are listed in terms of the reverse-complemented sequence.
Alignment Data Lines
Alignment data lines contain three required attribute values:
size dt dq
-
size-- the size of the ungapped alignment -
dt-- the difference between the end of this block and the beginning of the next block (reference/target sequence) -
dq-- the difference between the end of this block and the beginning of the next block (query sequence)
NOTE: The last line of the alignment section contains only one number: the ungapped alignment size of the last block.
"Snake" rearrangement display
Rearrangement display, sometimes called snakes display, is an alternative way to view pairwise alignments. It is available for PSL and chain format tracks.
Rearrangement display is a representation of the path that the sequence follows in the "other" sequence. You start in the upper left and move to the right, following the lines if you come to the end of a block. If a block is red, which means it is a match on the negative strand, then you reverse your course and start going from right to left. The gray lines mean there are no bases in the other sequence between the blocks. Orange lines means there are some bases in there that are not aligning.
The display type can be enabled on the track configuration page of eligible tracks. Below are two examples for clarity.
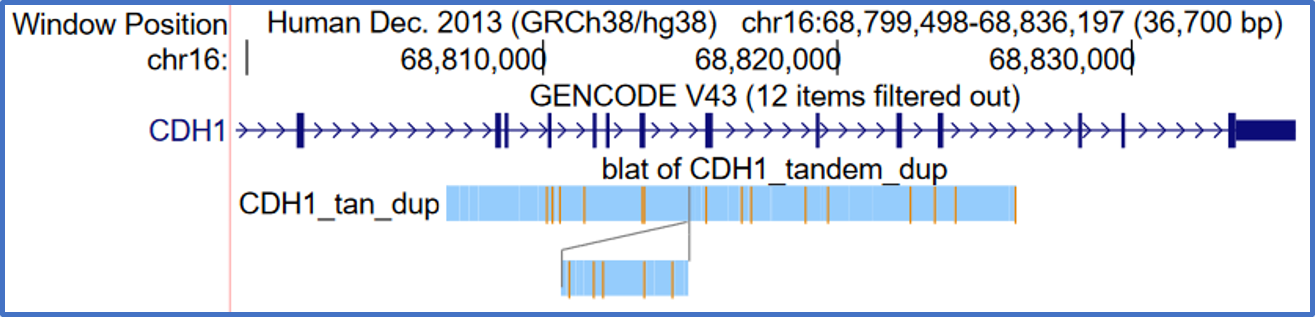
This example shows a tandem duplication on CDH1 which duplicates 3 exons.
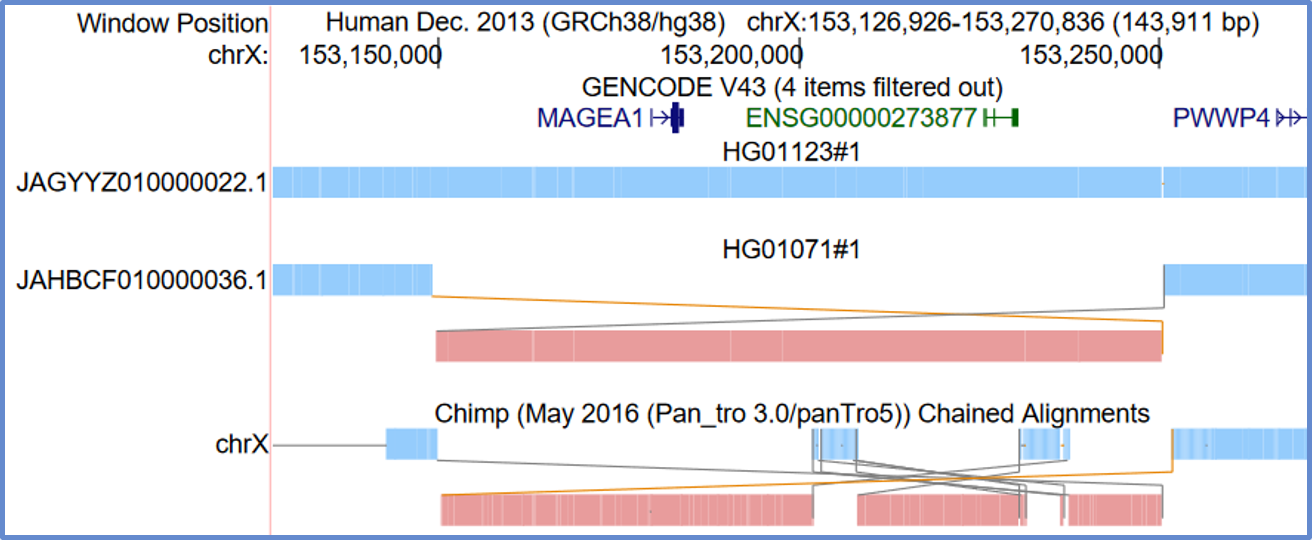
This example shows an inversion, which can be identified by the red colored sequence.